Department of Biosciences
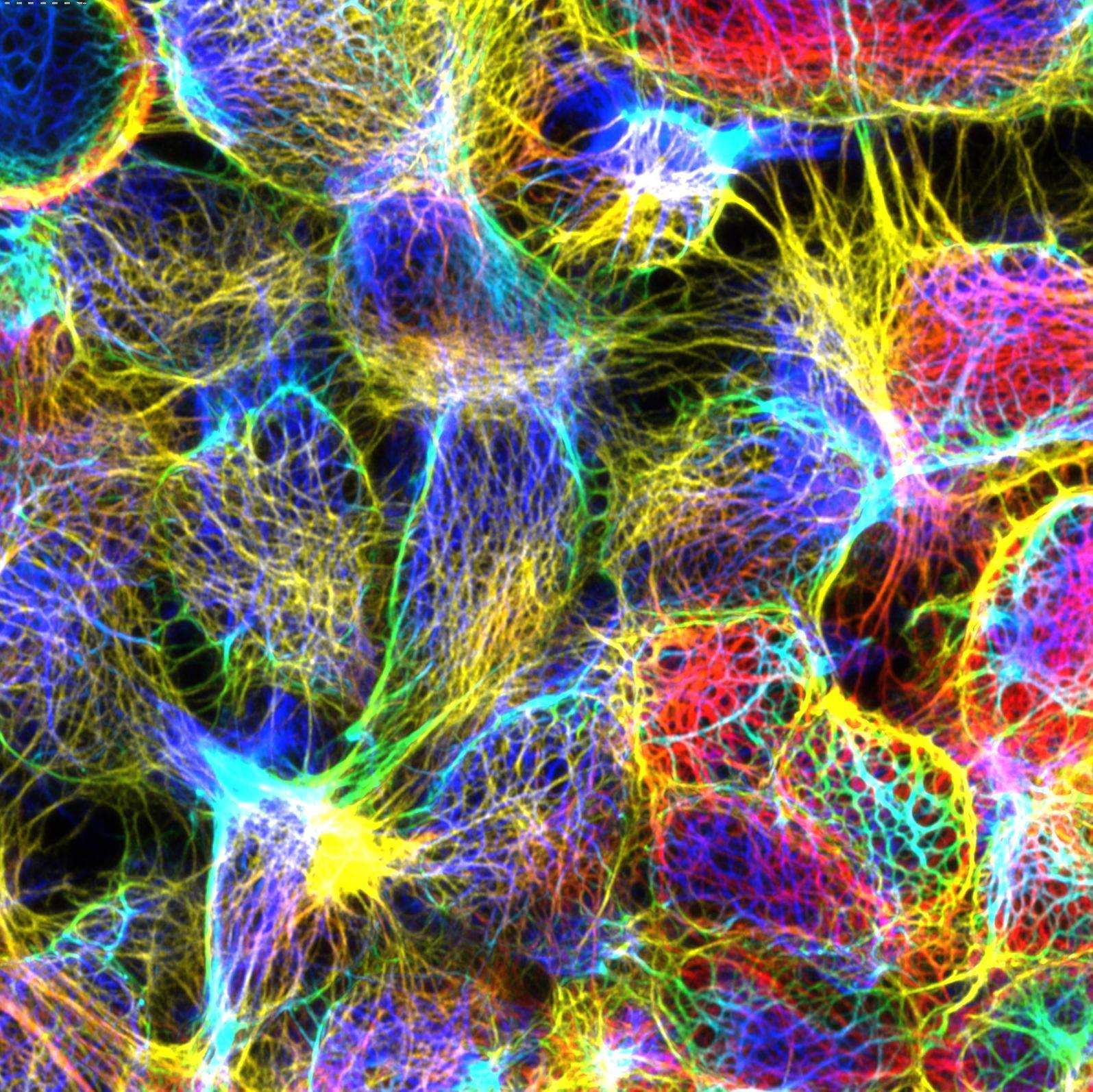

Welcome to the Department of Biosciences at Durham
Our research and teaching addresses fundamentally important questions facing humankind, from food security to sustainability in industrial processes, mechanisms of antimicrobial resistance and the impact of climate change on life on earth. Our students develop a wide range of analytical and practical skills that prepare them to meet these challenges.
Study with Us
-
Undergraduate Study
Our teaching is broad, from molecular structures to landscape-scale population dynamics, reflecting major questions in biology.

-
Postgraduate Study
Join a strong and vibrant postgraduate community for training in modern research techniques using state-of-the-art research infrastructure.
/prod01/prodbucket01/media/durham-university/departments-/computer-science/81569-1.jpg)
/prod01/prodbucket01/media/durham-university/departments-/biosciences/classroom/12257.jpg)
Undergraduate Study
Our teaching is broad, from molecular structures to landscape-scale population dynamics, reflecting major questions in biology.

Postgraduate Study
Join a strong and vibrant postgraduate community for training in modern research techniques using state-of-the-art research infrastructure.

Research
Discover more
/prod01/prodbucket01/media/durham-university/departments-/biosciences/infrastructure-/12479.jpg)
/prod01/prodbucket01/media/durham-university/departments-/biosciences/infrastructure-/12475.jpg)
/prod01/prodbucket01/media/durham-university/departments-/biosciences/12030.jpg)
/prod01/prodbucket01/media/durham-university/departments-/biosciences/infrastructure-/Crop-greenhouse.jpg)
News
-
Ancient origins of fallow deer should inform conservation
New research has revealed the hidden cultural histories of modern populations of fallow deer, dating back to the Neolithic period, which should be factored into decisions around their management and conservation.Research news/prod01/prodbucket01/media/durham-university/central-news-and-events-images/Fallow-Deer-WEB.png)
-
New study finds European breeding birds respond slowly to recent climate change
In a new study, leading scientists from our Department of Biosciences have found that local colonisation and extinction of European breeding birds are very weakly influenced by climate change.Research news/prod01/prodbucket01/media/durham-university/central-news-and-events-images/news/European-bird-WEB.png)
-
Strengthening and expanding protected areas of existing parks is crucial for biodiversity conservation
In a new study, bioscientists argue that strengthening the protection given to areas already protected under law or by local communities is as critical for safeguarding biodiversity as creating new protected areas.Research news/prod01/prodbucket01/media/durham-university/central-news-and-events-images/news/Protected-areas-species-WEB.png)
Ancient origins of fallow deer should inform conservation
New research has revealed the hidden cultural histories of modern populations of fallow deer, dating back to the Neolithic period, which should be factored into decisions around their management and conservation.

New study finds European breeding birds respond slowly to recent climate change

Strengthening and expanding protected areas of existing parks is crucial for biodiversity conservation

Upcoming events
At Durham, we provide an outstanding environment for research and education in animal, plant and bacterial biology encompassing expertise from the atomic to whole ecosystem scale. Trained by Durham Biosciences in the skills and knowledge required, our students are able to succeed in a wide range of careers or training environments and help society tackle the challenges for the future.
Work with us
We welcome new members to our team, bringing new approaches to answering biological questions.
Questions about studying here?
Check out our list of FAQs or submit an enquiry form.
Your Durham prospectus
Order your personalised prospectus and College guide here.